Hello everyone!
My name is Harout, a PhD student under the supervision of @ThomasTu and @MarkDouglas. The focus of my research project revolves around HBV subviral particles.
One part of my project involves manipulating the expression of a host gene, TM6SF2, to reduce/eliminate the secretion of HBV subviral particles.
Understanding the HBV Life Cycle
To begin, let’s briefly explore the replication and life cycle of HBV. Please refer to Figure 1 for this section:
Figure 1: HBV replication and life cycle
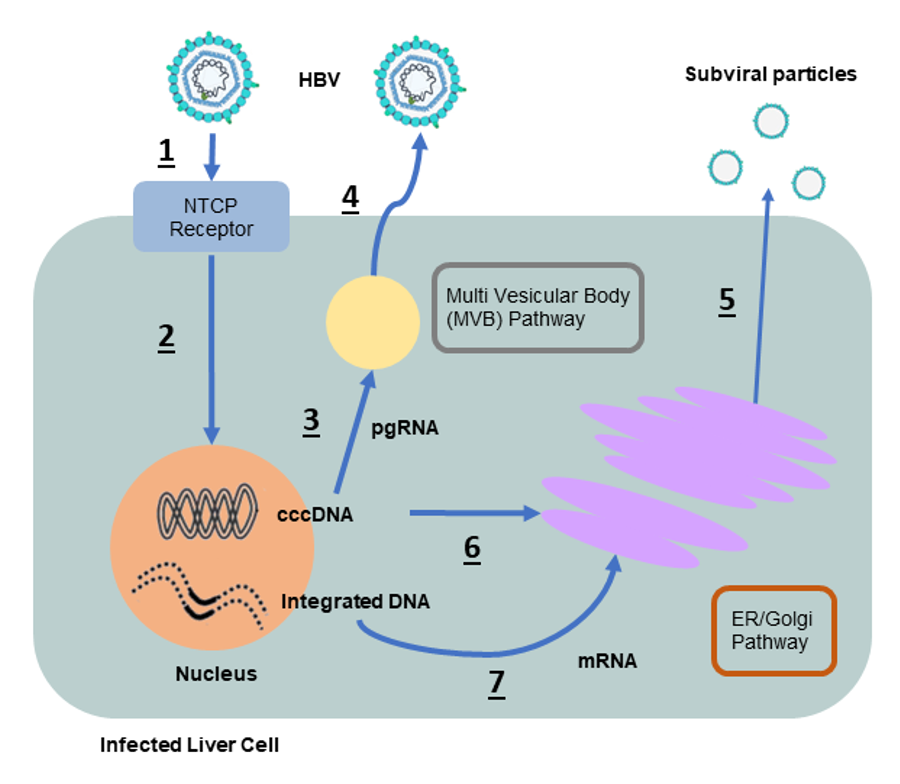
When HBV infects cells (path 1), it generates two forms of HBV DNA (path 2): cccDNA and integrated DNA. New viruses can only replicate from the cccDNA form (path 3). In addition to new viruses, chronic HBV infection is characterized by an excess of HBV subviral particles (path 5) with a ratio of about 100,000 subviral particles to 1 infectious virus.
These subviral particles are made of hepatitis B surface antigen (HBsAg) also known as the surface protein that envelops infectious viruses. Note: For the sake of simplicity, I’ll be calling subviral particles HBsAg from now on.
In the early stages of chronic infection, most of the HBsAg primarily originates from cccDNA (path 6) while in later stages, integrated HBV DNA becomes the major source (path 7).
The Challenge: Current Antiviral Drugs vs HBsAg
Unfortunately, current antiviral drugs (e.g. entecavir or tenofovir) have almost no impact on HBsAg. These drugs target viral replication at path 3 (Figure 1), leaving the production and secretion of HBsAg at paths 5-6-7 (Figure 1) completely unaffected. Consequently, even people undergoing antiviral therapy continue to produce substantial amounts of HBsAg.
Significance of HBsAg Elimination
Why do we care so much about HBsAg?
While HBsAg is non-infectious, it’s been shown to significantly interfere with the immune response against HBV by “distracting” and/or impairing the immune system. It is strongly believed that HBsAg is a major contributor to developing chronic HBV infection.
Consequently, the current medical guidelines for an HBV cure prioritize the achievement of a “functional” cure, which necessitates the clearance of HBsAg. As a result, multiple approaches to eliminate HBsAg are currently being explored.
This includes the one being investigated in our lab!
Our approach: TM6SF2
To eliminate HBsAg we are targeting the host gene transmembrane 6 superfamily 2, or TM6SF2. TM6SF2 normally controls the secretion of fats and cholesterol through the ER/Golgi secretory pathway. This happens to be the same pathway HBsAg is secreted from (see Figure 1).
Our group has previously shown that a mutation in TM6SF2 that reduces its levels by ~25% in the liver, also reduces circulating HBsAg levels by ~25% in people with chronic HBV. This finding prompted us to embark on a TM6SF2 project.
We silenced (or knocked down) TM6SF2 in cell culture (in vitro) models of HBV infection by small interfering RNA (siRNA) treatment, reducing its expression by ~85%. This resulted in a 50% reduction in secreted (extracellular) HBsAg levels!
Additionally, a significant increase in accumulated (intracellular) HBsAg was observed. Some recent studies have shown that treating HBsAg-accumulating cells with interferons can selectively target and eliminate them. Given HBsAg-accumulating cells are the cells infected with HBV, a co-treatment approach involving TM6SF2 and interferons could potentially pave the way for a complete cure of chronic HBV, including the elimination of cccDNA.
TM6SF2: A Promising Target
The most promising aspect of TM6SF2, is that it controls the downstream secretion pathway. Rather than targeting HBsAg production at paths 6 and 7 (Figure 1), TM6SF2 acts on path 5, effectively reducing secretion regardless of the source of HBsAg.
Our ultimate goal is to achieve efficient TM6SF2 knockdown-mediated HBsAg inhibition, allow the reactivation of the immune system, and pave the way for a HBV cure.
Thank you for taking the time to read my post. I’d be happy to answer any questions!