Hi all,
I’d like to share some ongoing work in my lab that is greatly improving our understanding of HBV replication and accelerating the drug discovery efforts in my lab.
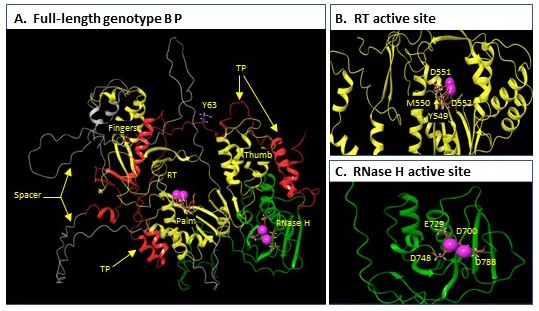
The HBV polymerase protein (abbreviated as “P”) is the only enzyme (ie, protein that makes chemical reactions occur) that HBV produces. It has 2 functions essential for HBV to copy itself, the reverse transcriptase (RT) that makes new viral DNA and the ribonuclease H (RH) that destroys the viral RNA after it has been copied into the first of 2 strands of the HBV DNA. Both are functions are essential for viral replication. The RT is the target of the nucleoside analog drugs like tenofovir and entecavir, but there are no drugs against the RH. Overall, our understanding of P’s function is extremely limited because the protein is incredibly difficult to work with.
I’ve been working with P since 1992 and have been making slow but steady progress. Over that entire time, I’ve wanted to know its 3 dimensional structure because a protein’s structure dictates its function. About 2 years ago Google figured out how to use artificial intelligence to accurately predict structures of most proteins, so we used their program (AlphaFold2) to predict the structure. We then worked really hard to validate that the prediction was right–and it is!. A free version of the paper reporting the structure is on bioRxiv at https://www.biorxiv.org/content/10.1101/2022.02.16.480762v1.
This structure has given us great ideas about how HBV P binds to the viral RNA, starts synthesizing DNA, and how the DNA and RNA move from the RT site to the RH site. This is interesting basic science in its own right, but it also has big implications about drug discovery. 1) it gives guidance to the drug companies how to make better nucleoside analog drugs; 2) it permitted us to launch multiple new drug discovery efforts against P that do not target the RT or RH sites (such drugs would be analogous to the NNRTI’s that work so well against HIV); and 3) it told us exactly why we were having so much trouble making active RH enzyme in the lab. We’ve used that information to engineer 2 versions of the HBV RH that can easily be made in the lab, and we are now adapting the enzyme to make assays that are useful for development of anti-RH drugs. Having such an enzyme in our toolbox is really accelerating our progress with the RH drug discovery project.
I’m personally deeply happy about this advance because I’ve been working on these problems for over 30 years, and they are finally beginning to crack. Hopefully they will yield benefits for HBV+ patients in time.
John