My name is Molly Woodson and I am a PhD graduate student in the lab of Dr. John Tavis at St Louis University, School of Medicine in St. Louis, MO USA. My main role in the lab is to assess the pharmacokinetic properties of novel HBV RNase H inhibitors.
The current treatment for chronic hepatitis B suppresses HBV well in most patients, but when treatment is stopped, viral replication rebounds. Therefore, the goal of our lab, and many others, is to identify therapies with new targets that can work together to decrease or stop the viral burden. The Tavis lab’s primary focus is on the HBV polymerase (P), which is responsible for viral DNA replication. One part of P, the reverse transcriptase domain, creates DNA from an RNA template; the nucleos(t)ide analogs inhibit this domain. The primary target of the Tavis lab is the ribonuclease H (RNase H) domain of P, which degrades the viral RNA. There are no drugs that currently target the RNase H despite it being an obvious drug target. This is because the assays needed for testing RNase H inhibitors were not available until our lab developed them over the last ten years.
Our lab collaborates with medicinal chemists around the world to synthesize and test novel HBV RNase H inhibitors, and recently I found “oxime HPD” compounds that inhibit HBV replication at very low concentrations with little to no toxicity. My current goal is to determine if these promising compounds are “druggable”. I determine this by screening the compound’s pharmacokinetic properties. Pharmacokinetics is broken down into four facets, and they are defined as absorption, metabolism, distribution, and excretion (ADME). These aspects answer the questions:
- Can my drug effectively get to where it needs to go (absorption)?
- Is my drug rapidly broken down by liver enzymes before getting to the site of action (metabolism)?
- Is my drug’s potency going to be decreased by binding to proteins in the blood (distribution)?
- Is my drug easily removed from the body (excretion)?
My research has determined that three promising HBV RNase H inhibitors are likely to be readily absorbed in the gastrointestinal tract, are unlikely to bind too much to blood proteins, and are not rapidly metabolized by liver enzymes (Figure 1). Additional aspects of my research determine if these promising inhibitors have off-target effects that are likely to result in increased toxicity or interactions with other drugs. I found that these three inhibitors are unlikely to inhibit or increase expression of the major drug metabolizing enzymes in the liver, and preferentially inhibit HBV RNase H over the human RNase H1 by 10-fold.
Thus far, these data indicate that the most effective oxime HPDs exhibit pharmacological characteristics similar to drugs on the market and are OK to move forward into advanced preclinical development as potential novel anti-HBV therapeutics.
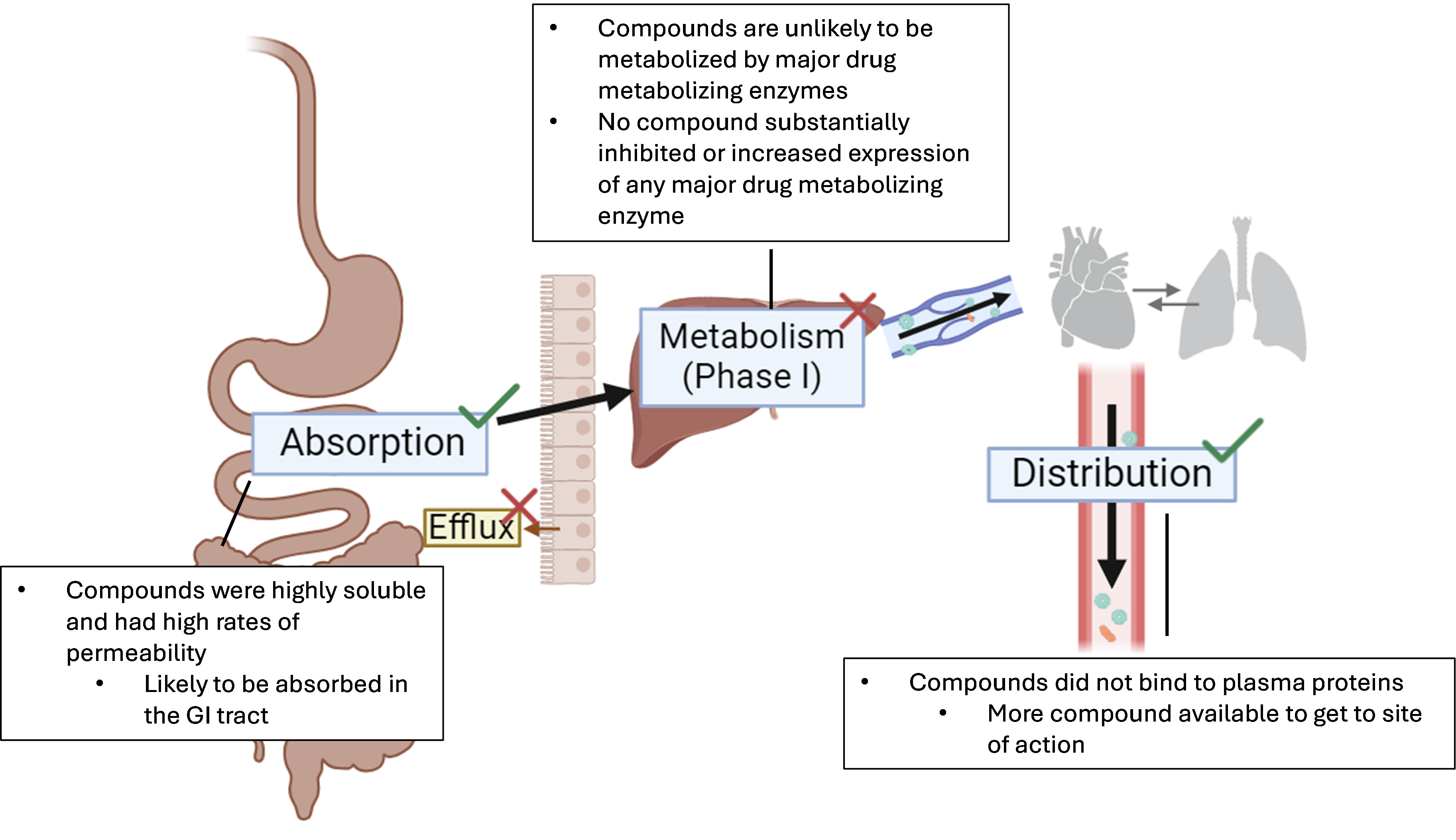
Figure 1. Absorption, metabolism, and distribution summary of 3 potent HPDs.